
Huntington Study Group (HSG) Conference 2024 –Dag1
Lees updates over klinische proeven en wetenschappelijk onderzoek naar de ziekte van Huntington, dag 1 van de 2024 Huntington Study Group-conferentie #HSG2024.
De Huntington Study Group (HSG) is een netwerk voor klinisch onderzoekklinisch onderzoek Zeer zorgvuldig geplande experimenten, ontworpen om specifieke vragen te beantwoorden omtrent het effect van een onderzoeksmiddel op mensen dat zich richt op het versnellen van behandelingen voor de ziekte van Huntington. Dit jaar wordt de jaarlijkse conferentie gehouden in Cincinnati, waar clinici, klinisch coördinatoren, maatschappelijk werkers, onderzoekers en farmaceutische bedrijven samenkomen om onderzoekupdates te delen en ideeën uit te wisselen. HDBuzz is aanwezig op de bijeenkomst en verzorgt live-tweets over de besproken onderwerpen. Voor wie onze live-updates heeft gemist, hebben we onze tweets gebundeld in een samenvatting. Lees verder om te ontdekken wat er gebeurde op Dag 1 van #HSG2024!
Welkom bij HSG 2024!
Het leiderschapsteam van de HSG opent de dag met een reeks introducties, een kort overzicht van de agenda, en een blik op de toekomst van de organisatie. Volg onze updates vandaag, en ook op vrijdag en zaterdag, voor het laatste nieuws over sessies rond klinische studies, gentherapie en innovatieve ontwikkelingen in het geneesmiddelenonderzoek.

MyHDStory
De daaropvolgende sessie omvat enkele beknopte updates over lopende klinische onderzoeken binnen de HSG. Dr. Karen Andersen van Georgetown University en Dr. Alex Dalrymple van de Universiteit van Virginia bespreken de eerste twee onderzoeksprojecten die worden uitgevoerd binnen het MyHDStory-platform.
MyHDStory is een online thuisstudie in Amerika, waaraan familieleden van mensen met de ziekte van Huntington individueel kunnen deelnemen. De sprekers leggen uit dat deze studie deelnemers de kans biedt om hun persoonlijke ervaringen met de ziekte te delen en zich aan te melden voor toekomstige online onderzoeken. Op deze manier kunnen zij een waardevolle bijdrage leveren aan de vooruitgang van huntington-onderzoek en de verbetering van zorg voor huntington-patiënten.
KINECT-HD
Dr. Erin Furr-Stimming van de Universiteit van Texas in Houston en Dr. Olga Klepitskaya van Neurocrine Biosciences presenteren vervolgens de bevindingen van de KINECT-HD-studie naar valbenazine. Dit medicijn werd begin 2024 in de Verenigde Staten goedgekeurd voor de behandeling van huntington-choreachorea Onvrijwillige, onregelmatige en ‘ongedurige’ bewegingen die veel voorkomen bij de ZvH, een symptoom van ongecontroleerde bewegingen bij de ziekte van Huntington (meer informatie: https://nl.hdbuzz.net/348 ).
De KINECT-HD-studie toonde overtuigend aan dat valbenazine effectief kan zijn in het verminderen van ongewenste bewegingen. Uit recente gegevensanalyses blijkt dat de meest voorkomende bijwerkingen, zoals episodes van slaperigheid, vooral in de eerste weken na het starten van het medicijn optreden en vervolgens geleidelijk afnemen.
Voor- en nadelen van huntingtine-verlaging: Een debat
Voordelen van huntintine-verlaging
De volgende sessie richt zich op de voor- en nadelen van huntingtine-verlagende therapieëntherapieën behandelingen. Dr. Blair Leavitt, klinisch huntington-onderzoeker aan de Universiteit van British Columbia, begint met een bespreking van de voordelen van deze aanpak.
Als achtergrond legt Dr. Leavitt uit dat extra CAG-herhalingen in het huntingtine-gen leiden tot een te lang eiwit. Uit onderzoek naar het menselijke gen en eiwit blijkt dat deze verlengde vorm van huntingtine toxisch kan zijn. Mensen met huntington die van nature minder verlengd huntingtine-eiwithuntingtine-eiwit Eiwit dat geproduceerd wordt door het huntington-gen produceren, ontwikkelen huntington-symptomen meestal op latere leeftijd, wat suggereert dat een lagere huntingtine-productie een beschermend effect kan hebben.
Dr. Leavitt deelt daarnaast gegevens uit onderzoeken met dier- en celmodellen, waaruit blijkt dat het verlagen van huntingtine vaak veilig en gunstig kan zijn. Er wordt echter nog steeds onderzocht wat de optimale mate van verlaging is om maximale voordelen te bereiken zonder bijwerkingen.
Hij benadrukt dat eerdere huntingtine-verlagende studies met tominersen (Roche), branaplam (Novartis) en Wave geen mislukkingen waren. Elk van deze onderzoeken heeft belangrijke inzichten opgeleverd. Onderzoekers passen nu de chemische samenstelling van deze geneesmiddelen aan, ontwikkelen nieuwe benaderingen voor het testen van therapieëntherapieën behandelingen en verwerken feedback uit de huntington-gemeenschap.
Nadelen van huntingtine-verlaging
In de daaropvolgende presentatie bespreekt Dr. Alberto Espay van de Universiteit van Cincinnati de uitdagingen en de mogelijke nadelen van huntingtine-verlagende therapieëntherapieën behandelingen. Hij benadrukt dat het huntingtine-eiwithuntingtine-eiwit Eiwit dat geproduceerd wordt door het huntington-gen in vele dierlijke soorten voorkomt en een rol speelt in verschillende cellen en organen, wat ons voorzichtig moet maken met een te grote vermindering ervan.
“De Huntington Study Group (HSG) is een klinisch onderzoeksnetwerk dat zich richt op het versnellen van de ontwikkeling van behandelingen voor de ziekte van Huntington . ”
Dr. Espay legt uit dat we weliswaar weten dat huntingtine normaal kan functioneren als het eiwit een gebruikelijke lengte heeft, maar dat we niet met zekerheid kunnen stellen dat het zijn functie verliest wanneer het verlengd is. Hij moedigt het publiek aan om om het als een grijs gebied te beschouwen ?, waarin een “abnormaal” eiwit zowel positieve als negatieve effecten kan hebben.
In de hersencellen van mensen met huntington vormen zich na verloop van tijd huntingtine-klonters, maar deze klontering leidt niet altijd direct tot celverlies. Daarnaast heeft huntingtine honderden interactiepartners, en niet alle wetenschappers zijn ervan overtuigd dat het verstandig is om die complexe interacties zomaar te onderbreken.
Hij wijst erop dat het definiëren van verlengd huntingtine als uitsluitend “toxisch” wellicht een te snelle conclusie is. Dr. Espay pleit ervoor om voorzichtig te zijn bij het bepalen wanneer en hoeveel huntingtine we willen verlagen.
Een alternatief kan zijn om de hoeveelheid “normaal” (ook “wildtype” ? genoemd) huntingtine te verhogen, in plaats van de verlengde, “mutante” vorm te verlagen. Dit idee werd eerder onderzocht in muismodellen van huntington.De effecten bleken niet overtuigend genoeg om verder te testen in klinische studies.
Gegevens zullen therapieëntherapieën behandelingen vooruithelpen
Deze levendige sessie is opgezet als een debat! Dr. Leavitt erkent het belang van huntingtine, maar weerlegt Dr. Espay’s suggestie om het wild-type huntingtine te verhogen. Hij legt de nadruk op het vele bewijs voor de giftigheid van de verlengde vorm. Extra wild-type eiwit toevoegen zal de toxiciteit van de verlengde vorm niet wegnemen. Wellicht kan men voor elk huntingtine-verlagend medicijn een therapeutisch raamwerk bepalen waarbinnen timing in combinatie met mate van verlaging, cruciaal zijn.
Dr. Espay spoort alle aanwezige bedrijven en onderzoekers aan om bij verdere innovaties rond huntingtine-verlaging rekening te houden met de belangrijke rol die het eiwit speelt in cellen.
Huntington-voorvechter en journalist Charles Sabine, OBE, neemt vervolgens het woord en herinnert iedereen eraan dat huntingtine-verlaging hoop blijft bieden, maar dat voor mensen met huntington ook andere onderzoeksstrategieën een goede toekomst kunnen bieden. Dr. Blair Leavitt sluit dit interessante debat krachtig af: “We zullen therapieëntherapieën behandelingen ontwikkelen op basis van gegevens!”
Medicijn afgifte
De volgende sessie behandelt de nieuwste ontwikkelingen in geneesmiddelenontwikkeling. Dr. Mali Jiang van Johns Hopkins werkt aan innovatieve afgiftesystemen voor huntingtine-verlagende en andere genetische therapieëntherapieën behandelingen. In het laboratorium van Dr. Lishan Lin richt Jiang’s werk zich op het herprogrammeren van levercellen om kleine “belletjes,” exosomen genaamd, te produceren. Deze exosomen kunnen genetische geneesmiddelen via de bloedbaan afleveren, wat een gerichte en efficiënte toediening via de bloedbaan mogelijk maakt.
Een belangrijke focus is het medicijn ER2001, dat reeds een kleine klinische test kende in China. Dr. Jiang presenteerde data over medicijnconcentraties in het lichaam na toediening via intraveneuze injecties over een periode van enkele maanden.
Het uiteindelijke doel van deze aanpak is om huntingtineverlaging te realiseren. Het Lin-laboratorium en het bedrijf dat dit onderzoek ondersteunt, ExoRNA Bioscience, hopen grotere studies (met ongeveer 30 deelnemers) op te zetten, zowel in China als in de Verenigde Staten, afhankelijk van de beschikbaarheid van financiële middelen.
PTC-518 heeft een nieuwe naam! Votoplam!
Vervolgens spreekt Brian Beers van PTC Therapeutics over de PIVOT-HD-studie naar PTC-518 dat voortaan votoplam heet. Eerder dit jaar schreven we over de veelbelovende vroege resultaten van deze studie. (https://nl.hdbuzz.net/370 ).
Brian vat de resultaten samen van onderzoek bij mensen die tot 12 maanden lang votoplam gebruikten. Deze deelnemers hadden lagere huntingtine-niveaus in hun bloed en hersenvocht dan degenen die een placeboplacebo Een namaakmedicijn zonder actieve ingrediënten. Het placebo-effect is een psychologisch effect waardoor mensen zich beter gaan voelen, zelfs als zij een pil innemen die niet werkt. (een pil zonder medicijn) kregen.

De studie heeft deelnemers gerekruteerd met een breed spectrum aan vroege huntington -symptomen. De gemelde bijwerkingen, zoals hoofdpijn, waren mild en een belangrijke update op deze conferentie is dat er geen reacties van het immuunsysteem zijn waargenomen. Zoals we in juni meedeelden, ziet PTC ook enkele vroege trends in verbeterde functionaliteit.
CAG-herhalingen controleren
De volgende sessie gaat over genbewerking (het aanpassen van DNA) en het tegengaan van somatische instabiliteit (het stoppen van CAG-herhalingsexpansie). Dr. Vanessa Wheeler van Mass General Hospital en Harvard Medical School gaf eerst een overzicht van recent onderzoek naar CAG-expansies op het niveau van individuele cellen. Deze techniek heeft de afgelopen tien jaar enorme vooruitgang geboekt en levert onderzoekers een grote hoeveelheden gegevens op. Vergelijk het met sterren kijken met het blote oog of met een krachtige telescoop – een wereld van verschil!
Vanessa bespr bespreekt ook GWAS-gegevens (genetische gegevens waarbij elk individueel gen van een persoon wordt geanalyseerd). Met deze grote datasets van mensen met het huntington -gen kunnen wetenschappers bepalen welke genetische informatie invloed heeft op de startleeftijd van symptomen.
Dit soort onderzoek identificeert genetische markers (modificatoren) die samenhangen met een eerdere of latere start van symptomen. Een beter begrip van deze modificerende genen helpt bij het vinden van mogelijke doelwitten voor medicijnen om de start van huntington-symptomen te vertragen.
Vanessa en haar team onderzoeken momenteel modificatoren van somatische instabiliteit – de voortdurende expansie van CAG-herhalingen in kwetsbare hersencellen. Ze hopen genen te identificeren die somatische instabiliteit controleren en die ze therapeutisch kunnen aanvallen.
Op dit moment testen ze deze doelgenen in huntington-muismodellen. Door CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken te gebruiken om de niveaus van deze doelwitten te wijzigen, ontdekten ze dat ze de hoeveelheid somatische instabiliteit kunnen controleren. Tot nu toe hebben ze 60 verschillende doelwitten getest. Dat is een enorm werk!
Elk modificator-gen dat somatische instabiliteit in cellen of muizen controleert, heeft de potentie om een therapeutisch doelwit te worden. Ze testen ook combinaties van doelwitten, wat behoorlijk ingewikkeld wordt met 60 verschillende doelen!
Vanessa en haar team houden er ook rekening mee dat individuele of gecombineerde doelwitten verschillende effecten kunnen hebben in verschillende celtypes. Zo kan een modificator die somatische instabiliteit vertraagt in ondersteunende hersencellen (glia), een ander effect hebben in neuronen.
Ze deelde daarnaast details over een specifieke modificator genaamd LIG1, die lijkt te zorgen voor minder somatische expansie in de hersenen van huntington-muizen. We wachten vol spanning op meer gegevens over deze modificator!
Vanessa’s uiteindelijke doel is om genetische modificatoren te identificeren die somatische expansie en instabiliteit controleren, hersencellen langer gezond houden, het begin van symptomen vertragen, en mensen met huntington meer gezonde en gelukkige jaren geven.
CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken voor genbewerking
Vervolgens bespreekt Dr. Ricardo Mouro Pinto van Mass General Hospital en Harvard Medical School de mogelijkheden van genbewerking in het onderzoek naar en de behandeling van de ziekte van Huntington (huntington).
Hij legt enthousiast uit hoe CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken – een krachtige genetische tool die werkt als een schaartje – specifieke DNA-sequenties kan ‘knippen’ en aanpassen. Onderzoekers kunnen hiermee DNA-doelen selecteren om ze te wijzigen, waarna CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken kan worden ingezet om die specifieke DNA-codes te corrigeren of te vervangen. Ricardo verwijst naar eerdere successen met CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken bij sikkelcelziekte als inspiratie voor huntington-onderzoek. (https://nl.huntington buzz.net/356)
Hij bespreekt vervolgens de technische details van verschillende manieren waarop specifieke DNA-basen kunnen worden aangepast, waarbij hij benadrukt hoezeer CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken in slechts tien jaar tijd is geëvolueerd.
“Elke gemodificeerde gen dat somatische instabiliteit in cellen of muizen reguleert, heeft het potentieel om een therapeutisch doelwit te worden.”
Twee belangrijke obstakels voor CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken bij huntington zijn echter de toediening en de veiligheid: hoe kunnen we CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken in de hersenen krijgen, en hoe zorgen we dat alleen de gewenste DNA-wijzigingen worden aangebracht?
Ricardo wijst op een recent financieringsinitiatief van de NIH (de Amerikaanse federale medische onderzoeksinstantie) voor de ontwikkeling van genbewerkingstherapieën, waarbij twee van de vijf gefinancierde projecten specifiek op huntington gericht zijn.
Een van deze projecten richt zich op het verwijderen van overtollige CAG-herhalingen in het huntingtine-gen, terwijl een ander project zich richt op het tegengaan van de somatische instabiliteit die tot steeds langere CAG-herhalingen kan leiden.
Interessant is dat sommige mensen met huntington een natuurlijke “onderbreking” in hun CAG-herhalingen hebben, waarbij een CAA de plaats inneemt van een CAG, wat vaak gepaard gaat met een latere aanvang van symptomen. Ricardo en zijn team werken aan technieken om CAG’s om te zetten naar CAA’s, wat mogelijk een beschermend effect zou kunnen bieden.
Daarnaast bouwt hij voort op eerdere presentaties over “modificatoren” van somatische instabiliteit, waarbij hij zich richt op het gen MLH3. Hij onderzoekt nieuwe CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken-technieken om MLH3 te verlagen, wat in cellen de groei van CAG-herhalingen kan vertragen of stoppen. De volgende stap is het testen van deze benadering in muizen en in menselijke cellen die huntington nauwkeuriger nabootsen.
Hoewel klinische proeven met mensen nog enige tijd op zich zullen laten wachten, is het bemoedigend om te zien dat dit werk aan steun en financiering wint. Het grootste voordeel van een potentiële genbewerkingstherapie is dat deze slechts één keer hoeft te worden toegepast om blijvende effecten te realiseren.
Huidige strategieën voor huntington-therapieëntherapieën behandelingen
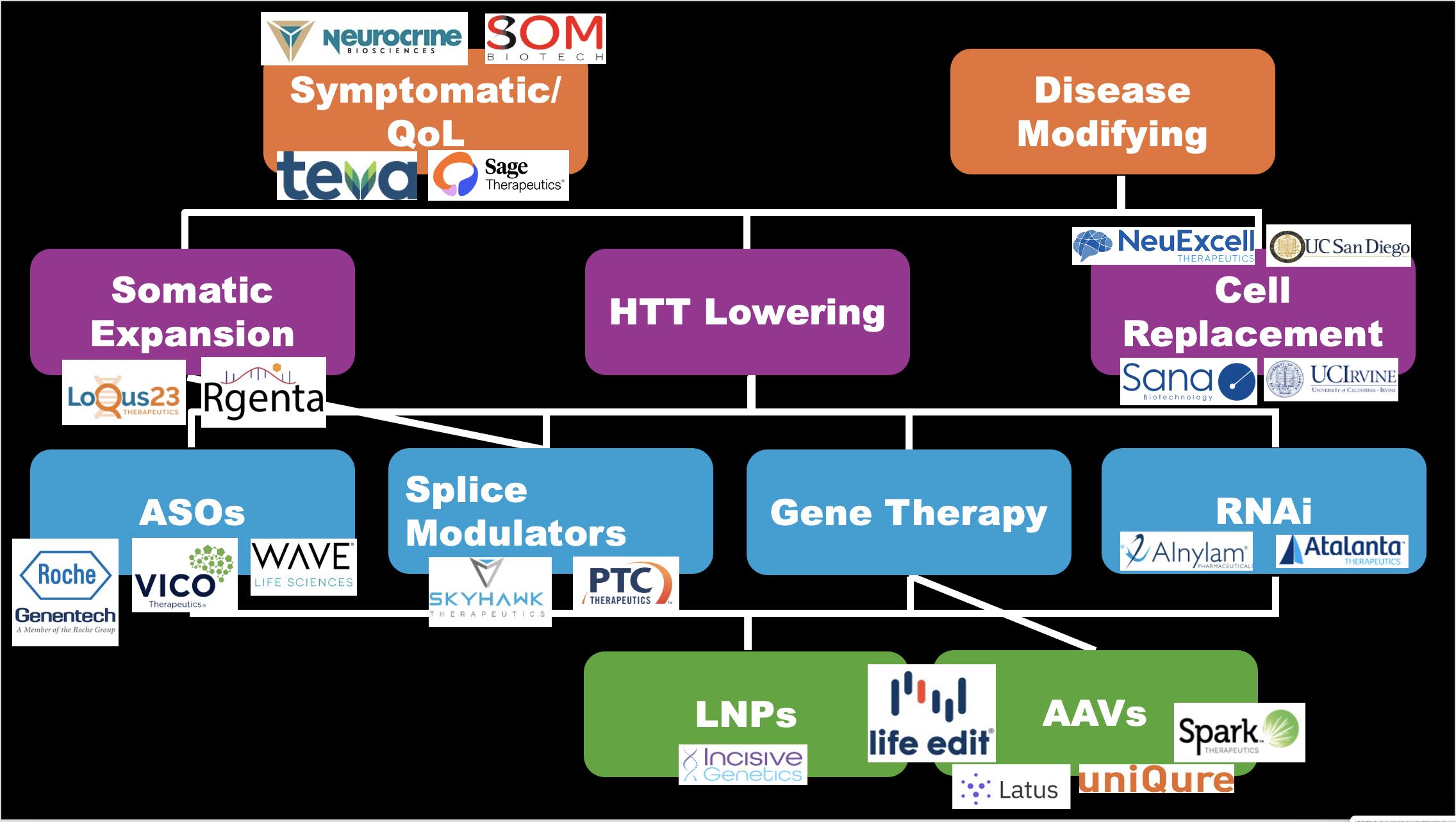
In de volgende sessie geven diverse bedrijven die werken aan innovatieve therapieëntherapieën behandelingen voor de ziekte van Huntington korte presentaties over hun laatste ontwikkelingen. Dr. Sarah Hernandez van HDBuzz start met een beknopt overzicht van de basisprincipes en de verschillende benaderingen die door bedrijven in dit veld worden gehanteerd.
Sarah bespreekt de uiteenlopende technieken en toedieningsmethoden die worden gebruikt om huntingtine te verlagen, zoals antisense-oligonucleotiden (ASO’s), RNARNA De chemische stof die lijkt op DNA en waaruit ‘boodschappermoleculen’ worden gemaakt. RNA wordt gebruikt als actieve kopie van genen bij de productie van eiwitten.-interferentie (RNAi), adeno-geassocieerde virussen (AAVAAV breingerelateerde neurotrofe factor: een groeifactor die mogelijk in staat is om neuronen (hersencellen) te beschermen bij de ZvH.’s) en splice-modulatoren. Elk van deze methoden biedt unieke mogelijkheden om de productie van het schadelijke verlengde huntingtine-eiwithuntingtine-eiwit Eiwit dat geproduceerd wordt door het huntington-gen te verminderen. Ze wijst erop dat de deelnemende bedrijven straks de gelegenheid krijgen om hun geneesmiddelenontwikkelingsprogramma’s in meer detail toe te lichten.
Daarnaast benoemt ze andere veelbelovende therapeutische strategieën voor huntington, zoals het tegengaan van somatische expansie van de CAG-herhalingen, het vervangen van verloren hersencellen, en het verbeteren van de communicatie tussen hersencellen. Deze benaderingen kunnen naast huntingtine-verlaging waardevolle aanvullingen bieden voor de behandeling van huntington-symptomen en ziekteprogressie.
Sarah sluit haar presentatie af met een indrukwekkende slide waarop de vele tientallen bedrijven te zien zijn die zich momenteel inzetten voor huntington-onderzoek. Ze benadrukt dat we ons in een opwindend tijdperk van klinische proeven voor huntington bevinden, wat hoop biedt voor mensen met huntington en hun families.
Alnylam Pharmaceuticals
De eerste spreker in het innovatorforum is Dr. Kevin Sloan van Alnylam Pharmaceuticals. Zij gebruiken een strategie genaamd RNAi – RNARNA De chemische stof die lijkt op DNA en waaruit ‘boodschappermoleculen’ worden gemaakt. RNA wordt gebruikt als actieve kopie van genen bij de productie van eiwitten.-interferentie – waarmee een stukje genetische code wordt toegevoegd om het huntingtinebericht te wijzigen en minder eiwit te produceren.
Kevin bespreekt eerst de details van de werking van RNAi. Een van de grootste uitdagingen voor RNAi-gebaseerde medicijnen is de levering/bezorging?. Ze willen ervoor zorgen dat het medicijn op de juiste plaats terechtkomt – wat voor HD betekent dat het zich verspreidt over het hele brein. Alnylam werkt ook aan andere ziektes, en Kevin deelt gegevens over een RNAi-medicijn voor de ziekte van Alzheimer dat nu de overgang naar een Fase 2-studie maakt.
Nu willen ze hetzelfde doen voor de ziekte van Huntington. Ze hebben een RNAi-medicijn ontwikkeld genaamd ALN-HTT02. Dit medicijn richt zich op alle vormen van huntingtine, inclusief korte stukjes die de neiging hebben om samen te klonteren. Deze worden huntingtine exon 1-fragmenten genoemd en men denkt dat ze giftig zijn voor hersencellen.
We hebben eerder geschreven over deze huntingtine exon 1-fragmenten, ook wel HTT1a genoemd, zie https://nl.hdbuzz.net/376 . Dit kleine fragment van het huntingtinebericht lijkt een eiwit te coderen dat alleen wordt geproduceerd bij mensen met het verlengde huntingtine-gen.
Alnylam test ALN-HTT02 nu bij apen, de stap voordat medicijnen naar klinische proeven met mensen gaan. Deze week kondigde Alnylam aan dat ze een Fase 1-studie starten voor dit medicijn! Deze eerste studie zal plaatsvinden in het Verenigd Koninkrijk en Canada, met rekrutering in andere landen die later gepland staat.
Het primaire doel zal veiligheid en verdraagbaarheid zijn, maar ze zullen ook kijken hoe goed het huntingtine aanvalt en hoe de niveaus veranderen in het hersenvocht (CSFCSF Heldere vloeistof geproduceerd door de hersenen die de hersenen en het ruggenmerg omringt en ondersteunt .), de vloeistof die de hersenen omgeeft. Ze zullen klinische tests gebruiken om symptomen te meten, maar een grotere studie is nodig om te bepalen of ALN-HTT02 de klinische kenmerken van HD daadwerkelijk verandert. Het is altijd spannend wanneer nieuwe studies worden aangekondigd, en we wachten vol spanning op nieuwe updates van Alnylam!
Rgenta Therapeutics
Als volgende spreekt Dr. Travis Wager van Rgenta Therapeutics. Zij ontwikkelen kleine moleculen die als pil kunnen worden ingenomen om somatische instabiliteit aan te pakken, de voortdurende toename van CAG-herhalingen in kwetsbare hersencellen.
Rgenta richt zich op een gen genaamd PMS1 (geen verband met stemming-gerelateerde PMS). De PMS1-niveaus zijn hoger bij mensen die vroeg symptomen van huntington vertonen. Rgenta probeert het PMS1-niveau te verlagen in de hoop het begin van huntington-symptomen te vertragen.
Travis legt uit hoe belangrijk het is om de juiste plaats op PMS1 te kiezen als doel. Ze hebben veel tijd besteed aan het ontwikkelen van moleculen die precies de juiste plek op PMS1 bereiken. De beste kandidaten voldoen ook aan andere criteria, zoals het kunnen doordringen in de hersenen wanneer ze als pil worden ingenomen.
Rgenta heeft het medicijn getest in verschillende diermodellen en aangetoond dat het zeer robuust is. Het verlagen van PMS1 met 50% vertraagt de instabiliteit met maar liefst 70% — de CAG-expansiesnelheid wordt aanzienlijk verminderd. Spannend nieuws!
Travis bedankte de onderzoekers binnen de huntington -gemeenschap voor hun samenwerking en het delen van middelen. Hij benadrukte dat Rgenta niet zou staan waar ze nu zijn zonder de open en samenwerkingsgerichte houding van huntington-wetenschappers.
Life edit
Dr. Logan Brown van LifeEdit Therapeutics presenteert de vooruitgang van hun baanbrekende gen-therapie voor de ziekte van Huntington, waarbij gebruik wordt gemaakt van CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken-technologie. LifeEdit werkt aan een CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken-gebaseerde behandeling genaamd LETI-101, die specifiek is ontworpen om alleen de verlengde (en potentieel schadelijke) versie van het huntingtine-eiwithuntingtine-eiwit Eiwit dat geproduceerd wordt door het huntington-gen te verlagen. Dit bereikt LifeEdit door zich te richten op een subtiel genetisch verschil dat vaak aanwezig is tussen de twee kopieën van het huntingtine-gen bij mensen.
LETI-101 kan echter niet bij iedereen met huntington worden toegepast. LifeEdit schat dat deze therapie effectief zou zijn voor ongeveer 30% van de mensen met het huntington-gen, omdat niet iedereen dit specifieke genetische verschil tussen de genkopieën heeft.
Het voordeel van deze benadering is dat, door CRISPRCRISPR systeem om DNA met grote nauwkeurigheid te bewerken voor genbewerking in te zetten, de therapie theoretisch als een eenmalige behandeling kan worden uitgevoerd, met levenslange verlaging van het huntingtine-eiwithuntingtine-eiwit Eiwit dat geproduceerd wordt door het huntington-gen als resultaat.
“We bevinden ons inderdaad in een tijdperk waarin klinische proeven een cruciale rol spelen in het onderzoek naar de ziekte van Huntington!”
Tot nu toe is LETI-101 met succes getest in celmodellen en muizen die huntington-symptomen vertonen. Momenteel wordt het medicijn in apen getest als voorbereiding op mogelijke klinische proeven met mensen. LifeEdit onderzoekt ook de praktische uitvoering van deze behandeling bij mensen, wat naar verwachting een chirurgische ingreep in de hersenen zal vereisen.
Sana Biotechnology
Dr. Joana Osorio van Sana Biotechnology gaf een update van hun baanbrekende werk aan celvervangingstherapieën voor de ziekte van Huntington, waarbij ze gebruik maken van stamcellenstamcellen cellen die kunnen delen in cellen van verschillende soorten, een cel die in staat is om in een ander celtype te veranderen (differentiëren). Sana richt zich specifiek op glia-cellen, de ondersteunende cellen in de hersenen die een rol spelen in huntington-gerelateerde neuronale problemen.
Experimenten toonden aan dat gezonde, niet-huntington glia-cellen, , symptomen van huntington wisten te verlichten wanneer ze in het brein van huntington-muizen werden getransplanteerd. De muizen vertoonden verbeteringen in hun motoriek, andere symptomen namen af, en hun levensduur werd verlengd.
Sana ontdekte dat de gezonde glia-cellen de zieke huntington-glia in de hersenen vervangen en hun functie gedeeltelijk overnemen. Dit geeft aan dat transplantatie van gezonde glia een potentieel effectieve aanpak zou kunnen zijn om de voortgang van de ziekte te vertragen of symptomen te verbeteren. Eerder heeft HDBuzz aandacht besteed aan deze veelbelovende resultaten, zie https://en.hdbuzz.net/347
Momenteel is Sana bezig met het vertalen van deze bevindingen naar een klinische context. de stap naar klinische tests kan nog niet meteen gezet worden, maar Sana werkt eraan om dit te realiseren. Het blijft een gebied om in de gaten te houden.
Spark Therapeutics
Dr. Juha Savola van Spark Therapeutics presenteert de vooruitgang van hun gentherapie voor de ziekte van Huntington, een project dat voortbouwt op Sparks eerdere successen met gentherapie voor erfelijk gezichtsverlies.
Spark ontwikkelt momenteel SPK-10001, een therapie die gericht is op het vertragen of mogelijk stoppen van huntington door de niveaus van het schadelijke huntingtine-eiwithuntingtine-eiwit Eiwit dat geproduceerd wordt door het huntington-gen (HTTHTT Afkorting voor het gen dat de ziekte van Huntington veroorzaakt. Hetzelfde gen wordt ook wel ZvH-gen of IT-15 genoemd.) te verlagen. In lopende dierstudies met apen heeft SPK-10001 veelbelovende resultaten laten zien: de HTTHTT Afkorting voor het gen dat de ziekte van Huntington veroorzaakt. Hetzelfde gen wordt ook wel ZvH-gen of IT-15 genoemd.-niveaus blijven tot wel 12 maanden verlaagd.
Bij de apen testen ze verschillende doses van SPK-10001 en volgen ze de verlaging van HTTHTT Afkorting voor het gen dat de ziekte van Huntington veroorzaakt. Hetzelfde gen wordt ook wel ZvH-gen of IT-15 genoemd. in verschillende hersengebieden. Dit helpt hen de juiste doses te kiezen om bij mensen te testen wanneer ze naar klinische proeven gaan. Juha deelt details over de inclusie- en exclusiecriteria voor de aankomende Fase I/II-studie die Spark plant bij mensen met de ziekte van Huntington. Deze studie zal twee doses testen: een lage en een hoge dosis.
Het primaire doel van deze studie zal veiligheid zijn, maar ze zullen ook enkele klinische parameters bekijken om een idee te krijgen of SPK-10001 effectief zou kunnen zijn in de behandeling van huntingtonsymptomen. We kijken vol verwachting uit naar de aankondiging van de rekrutering voor deze studie!
Atalanta Therapeutics
Dr. Serena Hung van Atalanta Therapeutics presenteert het werk van haar team aan ATL-101, een op RNARNA De chemische stof die lijkt op DNA en waaruit ‘boodschappermoleculen’ worden gemaakt. RNA wordt gebruikt als actieve kopie van genen bij de productie van eiwitten.-interferentie (RNAi) gebaseerde therapie die is ontworpen om de niveaus van het huntingtine-eiwithuntingtine-eiwit Eiwit dat geproduceerd wordt door het huntington-gen (HTTHTT Afkorting voor het gen dat de ziekte van Huntington veroorzaakt. Hetzelfde gen wordt ook wel ZvH-gen of IT-15 genoemd.) te verlagen bij de ziekte van Huntington.
Een van de unieke kenmerken van Atalanta’s aanpak is de geavanceerde leveringstechnologie die ATL-101 effectief naar diepe hersengebieden kan transporteren. Dit medicijn wordt toegediend via een ruggenprik en heeft aangetoond zich breed door de hersenen te kunnen verspreiden. In dierstudies bleek ATL-101 tot wel zes maanden actief te blijven.
Resultaten in apenstudies laten veelbelovende effecten zien: ATL-101 kan de HTTHTT Afkorting voor het gen dat de ziekte van Huntington veroorzaakt. Hetzelfde gen wordt ook wel ZvH-gen of IT-15 genoemd.-niveaus met maar liefst 75-90% verlagen. Bovendien bleven de niveaus van Neurofilament light (NfLNfL biomarker van gezondheid van hersencellen)stabiel – een marker die vaak toeneemt bij hersencelschade en ziekteprogressie. Dit behoud van NfLNfL biomarker van gezondheid van hersencellen-niveaus wijst erop dat ATL-101 mogelijk bescherming biedt aan hersencellen en het verloop van huntington zou kunnen vertragen.
Atalanta Therapeutics is van plan om in 2025 met klinische proeven voor ATL-101 te beginnen. Dit is boeiende research, wij houden jullie op de hoogte.
Wees morgen terug present
We gaan dan verder met het verkennen van meer dan 80 onderzoeksposters die gepresenteerd worden door huntington-wetenschappers en clinici van over de hele wereld. Korte presentaties zullen licht werpen op biomarkers voor hersenbeeldvorming en aspecten van het Enroll-HD platform. Tot morgen voor sessies over klinische uitdagingen bij huntington, biomarkers, trialontwerp, en meer.
For more information about our disclosure policy see our FAQ…


